Pourquoi créer une banque d’insertion pour trouver des mutants plutôt que réaliser une mutagenèse aléatoire par UV (comme dans le TD 2)?
Une mutagenèse aléatoire par UV permet de créer
toutes sortes de mutations : changement d’un a.a. dans un
peptide, modification du niveau d’expression génique,
abolition d’un gène (Knock-Out, KO), etc. Par
conséquent, il y a un très grand nombre de possibilités
de trouver la mutation qui vous donnera le phénotype
recherché. Son principal désavantage par contre, c’est
que vous ignorez totalement où se trouve la mutation sur le
génome de votre organisme. L’identification va par
conséquent être fastidieuse.
Au contraire, une
banque d’insertion est composée de fragments issus du
génome, dans lesquels a été inséré
un marqueur. Par conséquent, les seules mutations que vous
observerez seront des KO (et éventuellement des modifications
du niveau d’expression génique). L’avantage vient
du fait que vous connaissez la séquence du marqueur inséré
et, par conséquent, il est très facile de séquencer
la région (locus) dans laquelle le marqueur est inséré.
L’identification du gène « inactivé »
est donc très facile.
Quels sont les inconvénients que peut présenter une banque d’insertion?
Comme nous l’avons vu à la question précédente,
une banque d’insertion ne permet que d’inactiver un gène
(KO). Il n’est pas possible de réaliser des mutations «
subtiles » (changement d’un a.a. dans une protéine)
par insertion d’un marqueur. Par conséquent, vous
passerez peut-être à côté de nombreux gènes
impliqués subtilement dans le processus que vous étudiez.
Un
autre inconvénient peut venir de votre méthode
d’insertion. Certains transposons ne s’intègre
qu’à des sites spécifiques. Par conséquent,
il est possible que vous ne puissiez pas effectuer un KO de tous les
gènes de l’organisme.
Un ORF de 999pb code pour un peptide de 332aa. Pourquoi pas 333 ?
Le code génétique utilise un système de codon de 3nt pour chaque acide aminé. Un ORF (Open Reading Frame, ou Cadre de Lecture Ouverte, en français) commence par un codon Start (AUG, qui code pour une Méthionine) et s’achève par un codon Stop (UAA, UAG, UGA ; qui ne code pour aucun acide aminé). Il faut donc enlever le codon stop de votre séquence pour connaître la longueur théorique (en a.a.) de votre peptide. 999-3 = 996. 996/3 = 332 a.a.
C. albicans ne croît que sous forme diploïde asexuée. Pourquoi cela pose-t-il un problème pour des recherches génétiques ?
Pour une cellule, l’avantage d’avoir 2 copies de
chaque gène est un avantage certain. La plupart des mutations
inactivant une des copies laissera l’autre fonctionnelle. La
cellule se portera donc probablement aussi bien que si elle avait ses
2 copies fonctionnelles. La plupart des mutations produites par
insertions sont récessives (attention, il y a toujours des
exceptions). Il faut donc inactiver les 2 copies pour exprimer le
phénotype mutant, ce qui, par conséquent, pose un
problème technique pour vos expériences. Vous devez
inactiver les deux copies (une par chromosome homologue). Il vous
faudra donc 2 marqueurs différents de sélection,
insérés dans le même gène mais chacun sur
un des deux chromosomes homologues.
La méthode utilisée
par les chercheurs ayant réalisé cette expérience
est très belle. Mais, cela sortant très largement du
cadre de ce TD, nous vous renvoyons donc à la lecture des
articles cités, pour
les plus intéressés.
Pourquoi est-il important qu’une banque génomique couvre plus d’une fois le génome ?
En coupant l’ADN aléatoirement par une enzyme de
restriction, vous risquez de couper à l’intérieur
d’un gène. Par conséquent, les fragments
retrouvés ne contiendront pas toujours des gènes
entiers. Il vous faut donc couper plusieurs fois le génome
entier de l’organisme étudié, à différents
endroits, pour vous assurer d’avoir au moins 1 fois chaque gène
de l’organisme (en entier), contenu dans votre banque.
De
plus, en ayant qu’une seule copie de chaque gène dans
votre banque, vous prenez le risque de perdre certains gènes
durant les phases d’amplification de votre banque. Il vous est
donc nécessaire d’avoir plus d’une copie de chaque
gène pour être certain d’avoir une banque
génomique complète jusqu’à la fin de
l’expérience.
Quel est l’intérêt d’avoir des fragments (de la banque génomique) chevauchants ?
Cette question trouve sa réponse dans la précédente. Comme vous désirez au moins une fois chaque gène de l’organisme en entier dans votre banque, il vous faudra effectuer une digestion partielle de plusieurs génomes complets, ce qui vous donnera obligatoirement des fragments chevauchants. L’intérêt d’avoir des fragments chevauchants est de vous assurer de toujours avoir l’ensemble d’un gène dans votre banque (promoteur, terminateur, ORF et régions régulatrices !). Par exemple, si un fragment contient les gènes A,B et partiellement C, un autre fragment contiendra probablement les gènes B (partiellement), C et D (entièrement).
Voyez-vous d’autres moyens de procéder pour fabriquer une collection de mutations par insertion ?
On peut imaginer (en théorie c’est possible, en pratique cela ne se fait pratiquement pas) une insertion réalisée par restriction puis ligation d’un marqueur, dans votre ADN génomique. Il est également possible de faire une PCR du marqueur avec des amorces contenant des séquences d’homologies pour chaque gène (il y aurait donc autant de couples d’amorces différents et de PCR, que de gènes) et d’intégrer ces fragments par recombinaison in vivo. Il existe d’ailleurs, pour certains organismes, des collections de souches KO, que vous pouvez acheter.
Si vous désirez construire une banque génomique afin d’effectuer une complémentation, auriez-vous fait le même choix de plasmide?
Absolument pas. Pour effectuer une complémentation avec un vecteur contentant un fragment génomique, vous devez vous assurer que le vecteur peut être répliqué efficacement dans la cellule cible (ici C. albicans). Par conséquent, il est nécessaire d’avoir les séquences d’initiation de réplication adaptées à l’organisme étudié.
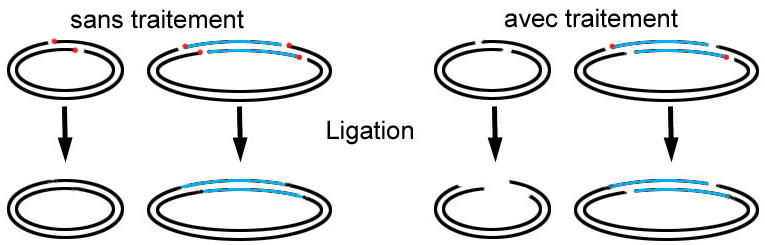
Les bouts francs d’ADN sont plus difficiles à liguer, pourquoi ?
L’avantage des bouts cohésifs est qu’il y a un appariement (hybridation) de quelques bases entre vos deux fragments devant être ligué. Par conséquent, lors de la rencontre des fragments, l’appariement des bases entre-elles maintient les fragments ensemble, un peu plus longtemps que s’il n’y avait pas d’appariement (ce qui est le cas des bouts francs). La probabilité de réaliser une ligation est donc d’autant plus importante que la longueur du bout simple brin dépassant est grande.
Dans ce cas, pourquoi y a-t-il tout de même ligation avec un vecteur traité et un fragment non-traité ?
Comme vous le remarquez sur le schéma, le fragment génomique n’ayant pas subi de traitement, possède toujours ses phosphates en 5’. Il y a donc ligation d’un seul brin à chaque site de restriction, ce qui est largement suffisant. Une fois dans la bactérie, le «trou» restant sera réparé.
Le système lacZ, comment ça fonctionne ?

Voici justement la démonstration de la mutation par insertion. Le gène lacZ code pour une enzyme qui dégrade le lactose en galactose et glucose. Cette enzyme peut également couper un composé chimique, le X-Gal, qui, une fois coupé, devient bleu. Si un fragment génomique s’est inséré dans l’ORF de lacZ, il a toutes les chances d’abolir le cadre de lecture de ce dernier et donc d’abolir la production d’une enzyme fonctionnelle. Sans enzyme, il n’y aura pas de clivage du X-Gal et la bactérie restera blanche.
Pour une banque de complémentation, vous avez besoin de fragments de 3 à 8 Kb, pourquoi ?
Pour une banque de complémentation, vous souhaitez, en
général, disposer d’un seul gène par
fragment génomique, ce qui vous facilitera l’identification
de votre mutation originale. En effet, si vous avez 3 gènes
sur un plasmide supprimant le phénotype mutant étudié,
vous ne saurez pas lequel des trois est le gène
suppresseur.
Si la longueur moyenne théorique d'un gène
de C. albicans est de 2.3 kb (ORF d'environ 1.5 kb), alors il
vous faudra des fragments entre 3kb et 8kb.
Pourquoi ajoutez-vous du glycérol et un colorant à vos échantillons, avant de les charger sur le gel d’électrophorèse ?
Pour effectuer une bonne migration, il faut que le courant
électrique puisse passer entre les électrodes. Ceci est
assuré par la présence d’un tampon salin qui
recouvre le gel. Si vous placez un liquide dans une solution, il va
diffuser à partir de l’endroit où vous l’avez
placé (ici le puits du gel). En ajoutant le glycérol,
votre échantillon sera plus visqueux et plus dense que le
reste du solvant. Par conséquent il va tomber au fond du puits
et diffuser bien plus lentement.
Quant au colorant, c’est
simplement un repère visuel pour vous montrer où se
trouve vos échantillons. Cela vous évitera de mettre
involontairement deux échantillons dans le même puits,
et cela vous permettra également de contrôler
l’avancement de votre migration (à condition que le
colorant soit chargé et migre librement dans le gel).
Pensez-vous que ce type de visualisation (EtBr et UV) peut causer du tort à votre expérience ? Pourquoi, comment ?
Evidemment que oui. Vous avez vu dans le TD2 les dommages causés par les UV sur l’ADN. Quant au bromure d’éthydium, comme il s’intercale entre les bases, il peut causer des mutations durant la réplication de l’ADN. Cependant, comme on s’assure de l’enlever avant de transformer les bactéries avec ces ADN purifiés, il ne devrait pas y avoir de problème de ce côté là. En résumé, vous ne devez pas laisser trop longtemps vos ADN sous la lampe UV.
La méthode consiste à faire fondre le gel d’agarose à 50°C au maximum, pourquoi pas plus de 50°C ?
Vous avez vu durant le TD3 (PCR) qu’en chauffant les ADN, on pouvait les dénaturer. Comme vous ne souhaitez pas effectuer de dénaturation, vous évitez de chauffer au delà de 50°C. Evidemment le Tm de vos fragments est probablement bien plus élevé que 50°C. Cependant, je vous rappelle que le Tm correspond à la température pour laquelle 50% des fragments sont dénaturés. 50% ici, c’est déjà trop.
La ligase a besoin d’ATP pour coller les fragments d’ADN, pourquoi ?
La formation d’une liaison phosphodiester nécessite de l’énergie. Durant la polymérisation de l’ADN au cours de la réplication, cette énergie est fournie directement par le nouveau nucléotide (qui est tri-phosphate et donc hautement énergétique). Par contre, durant la ligation, vous n’avez plus qu’un nucléotide monophosphate et donc pauvre en énergie. L’enzyme ligase se sert de l’énergie contenue dans la molécule d’ATP pour former la liaison phosphodiester sur votre brin d’ADN.
Comment allez-vous faire la différence entre une bactérie ayant reçu un plasmide avec un fragment d’une bactérie n’ayant rien reçu, ou ayant reçu un plasmide/fragment linéarisé (non ligué) ?

Un fragment seul ne contient pas d’origine de réplication.
Il ne peut donc pas être répliqué durant la
division de la bactérie et ne sera donc pas amplifié.
Un
vecteur non ligué ne pourra pas non plus être répliqué
efficacement. En effet, souvenez-vous de l’importance des
télomères et de la télomérase, dans la
maintenance des chromosomes linéaires. Les bactéries ne
possèdent pas de télomérase. De plus (et
surtout, en fait), les extrémités de votre vecteur ne
sont pas protégées contre les nucléases de la
bactérie et votre plasmide sera donc dégradé.
En
résumé, seuls les plasmides ayant reçu un
fragment seront amplifiés dans les bactéries. Pour
différentier les bactéries ayant un plasmide de celles
n’en ayant pas, vous allez utilisé le gène de
résistance à un antibiotique contenu par votre
plasmide. Ainsi, en ajoutant l’antibiotique à votre
milieu de culture, seules les bactéries avec un plasmide
pousseront.
Pourquoi ne pas réaliser plutôt l’amplification d’ADN par PCR, comme dans le TD 3 ?
Fondamentalement c’est possible. Cependant, en terme de ressources et de temps utilisés, c’est impossible de procéder par PCR. En effet, vous avez beaucoup de fragments différents. Comme le temps d’élongation d’une PCR doit être calculé en fonction de la longueur du fragment à amplifier, cela vous pose un premier problème. Ici, les fragments sont très longs, ce qui signifie réaliser des PCR terriblement compliquées (plus le fragment est grand et plus la PCR est difficile) et longues.
Est-ce que toutes les bactéries transformées portent le même plasmide avec le même fragment ? Si non, vous est-il utile de trier chaque clone séparément, plutôt que de cultiver vos bactéries toutes ensemble ?
Non et non.
Vous établissez une banque, par conséquent
vous avez beaucoup de plasmides contenant des fragments génomiques
différents. Chaque bactérie transformée aura
donc un plasmide différent. Toutes les bactéries issues
(colonie) de cette bactérie transformée, auront par
contre le même plasmide.
Comme vous désirez une
banque, vous souhaitez garder vos fragments ensemble, il est donc
inutile de les trier séparément. Sans oublier que si
vous deviez conserver chaque clone indépendamment, cela
représenterait facilement 10'000 tubes différents !
S. mutans est anaérobe et aérotolérante, quelle différence entre ces 2 termes ?
On peut classer les organismes en fonction de leur besoin et de leur tolérance à l’oxygène (O2). Une bactérie anaérobe signifie qu’elle n’utilise pas l’O2 pour sa production d’énergie. Au contraire, une bactérie aérobe utilise l’oxygène pour produire son énergie. La tolérance, elle, représente le fait que la bactérie supporte la présence (anaérobe aérotolérante) ou non (anaérobe stricte) de l’oxygène. L’inverse est également vrai pour les bactéries aérobes (nous autres humains, sommes aérobes strictes).
S. mutans produit son énergie à partir de la dégradation du glucose en acide lactique. Voyez-vous un lien avec la formation de caries ?
J’espère évidemment que oui. En mangeant du sucre, vous nourrissez les bactéries, qui produisent de l’acide lactique. L’acide produit fait baisser le pH à la surface de vos dents, ce qui détruit la couche d’émail qui les protège. La couche d’émail détruite, les bactéries peuvent infiltrer le reste de la dent… et vous commencez à souffrir.
Voyez-vous une différence stratégique entre l’utilisation du 14C et celle du 35S ? Sachant qu’il n’y a pas de grandes différences d’intensités de radiations produites par ces deux isotopes.
La principale différence réside dans les acides aminés qui peuvent être marqués. Le souffre ne se trouve que dans deux acides aminés : Cystéine et Méthionine. Le carbone, en revanche, se trouve dans tous les acides aminés (et pas qu’eux d’ailleurs). La Cystéine étant un a.a. relativement rare dans les protéines, si vous l’utilisez comme marqueur radioactif, vous aurez un marquage spécifique des protéines qui la contiennent.
Vous disposez donc de 4 cultures différentes. Pourquoi en faire autant?
Si vous désirez observer les changements entre deux conditions, il vous faut évidemment réaliser l’expérience pour chaque condition. Comme vous étudiez ici quatre conditions (présence ou non d’acide et biofilm on non) cela vous fait donc 4 cultures.
Quelles mesures de protection devez-vous prendre avec la radioactivité?
Portez des vêtements de protection (blouse, gants, etc).
Utilisez un écran plombé qui stoppe les rayonnements
issus de la fission des atomes. Portez un badge de mesure (qui
enregistre la somme des radiations que vous avez subie) et
munissez-vous d’un compteur Geiger (qui permet de détecter
la plupart des radiations). Travaillez proprement et vérifiez
vos surfaces de travail avant et après utilisation de la
radioactivité. Evitez d’exposer vos collègues aux
radiations (l’écran de protection n’est pas que
pour vous).
Il est évident qu’il est peu probable que
vous vous retrouviez un jour à manipuler de la radioactivité.
Cependant, il est fort probable que vous manipuliez un jour du
matériel sanguin contaminé par un pathogène
particulièrement dangereux. Pathogène et radioactivité
se ressemblent sur 2 points : ils sont dangereux et sont invisibles à
l’oeil nu. La plupart des règles de prudence sont donc
valables dans les deux cas.
Le renouvellement des protéines dans les bactéries est assez important. Est-ce la même chose pour les protéines humaines p.ex ?
La plupart des bactéries ont une croissance relativement rapide, il leur est nécessaire de fabriquer beaucoup de protéines. Les bactéries (comme les autres cellules d’ailleurs) ont souvent besoin d’une protéine donnée uniquement à un moment précis. Par conséquent, beaucoup de protéines sont activement dégradées une fois leur fonction remplie. Il y a donc un renouvellement important des protéines dans une bactérie. Les cellules eucaryotes ont souvent un rythme de croissance plus lent. La plupart des protéines de ces cellules ont donc une durée de vie plus importante. Cependant il y a des exceptions dans les deux royaumes.
Est-ce que deux protéines composées des mêmes acides aminés peuvent avoir un pI différent ?
Oui. Les protéines sont souvent modifiées après la traduction : elles subissent des phosphorylations, des glycosylations, etc. Ces modifications sont rarement neutres sur la charge nette d’une protéine.
Est-ce que des protéines ayant le même pI peuvent avoir des PM différents ?
Oui. Le pI dépend de la charge nette des protéines; par conséquent, des compositions différentes en a.a. peuvent conduire au même pI, et ce indépendamment du nombre d’a.a (et donc du PM) de vos protéines.
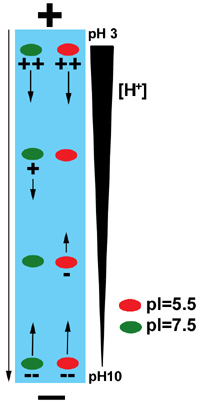
Auriez-vous pu charger vos protéines à l’autre extrémité ?
Oui. Comme illustré sur l’image qui suit :
Pourquoi n’avoir pas rajouté le SDS avant l’isoélectrofocalisation ?
En ajoutant le SDS avant l’isoélectrofocalisation, vous auriez modifié la charge nette des protéines, rendant ainsi impossible une séparation selon le pI de chaque protéine.
Au fait, vers quel pôle migrent les protéines liées au SDS ?
Vers le pôle +. Le SDS est chargé négativement. Par conséquent, les protéines fixant le SDS acquièrent une charge nette négative.
Durant l’isoélectrofocalisation, de quel côté du gel avez-vous branché le pôle positif ?
Le + du côté acide (pH bas). En milieu acide, les protéines sont chargées positivement, et vont donc migrer vers le pôle négatif (que vous devez donc placer à l’autre extrémité).
Connaissez-vous l’examen utilisant des isotopes radioactifs ?
Il s’agit de la scintigraphie. Cet examen clinique consiste à injecter un produit légèrement radioactif, considéré comme un traceur et qui va être distribué dans l’organisme en fonction de sa nature (chimique, et pas radioactive). A l’aide d’un gros détecteur, vous pourrez observer la position de l’élément radioactif dans le corps. Voici un lien définissant la scintigraphie.
Est-ce que le principe de la radiographie est le même que celui de l'autoradiographie ?
Non. Dans l’autoradiographie (principe utilisé par la scintigraphie), l’émission des rayons détectés provient d’une source située dans l’objet observé (ou le patient). Dans la radiographie, la source est extérieure à l’objet (ou le patient) et les rayons le traversent avant de frapper le détecteur.
Souvenez-vous du type de dommages créés par les radiations ionisantes sur l’ADN ?
Il existe plusieurs types de dommages causés par les
radiations ionisantes (rayons de haute énergie : rayons X,
rayons gamma). Il y a notamment les ruptures des liaisons
phosphodiester provoquant des cassures double-brins de l’ADN.
Ces cassures sont normalement réparées par la cellule
grâce à la recombinaison homologue, mais si ces cassures
sont trop nombreuses, la réparation devient impossible.
Les
cellules les plus affectées par ces rayons, sont les cellules
en division. Les rayons X sont, par conséquent, utilisés
pour traiter certains cancers (radiothérapie). Et si vous avez
déjà subi un examen radiographique, vous aurez
peut-être remarqué que le radiologue aura placé
un tablier en plomb afin de protéger vos gonades (organes
reproducteurs primaires).
Combien de protéines différentes trouve-t-on dans une bactérie ?
Tout dépend de la bactérie, de son cycle cellulaire et des conditions de son environnement. Etant donné qu’une bactérie possède entre 2000 et 5000 gènes codant pour des protéines, il est raisonnable d’imaginer que la bactérie utilise entre 500 et 2000 protéines différentes à un moment précis.
On peut identifier le gène qui code pour cette protéine à partir de la séquence de la protéine et de la séquence du génome. Comment est-ce possible ?
Le séquençage du génome nous permet de
connaître la grande majorité des ORF (ce qui est très
facile dans les organismes ne contenant que très peu
d’introns, et nettement plus difficile dans les autres). Grâce
au code génétique, qui est respecté presque
universellement (il existe des exceptions au code), on peut déduire
la séquence de la protéine codée par cet ORF.
Pour retrouver le gène correspondant à une
protéine, il suffit alors de comparer la séquence
obtenue par séquençage de la protéine avec les
séquences déduites des ORF identifiés dans le
génome de l’organisme.
B. Emery. 21.11.05