Quel additif utiliser ?
|
|
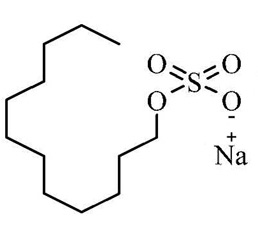
Bonne réponse. Le SDS.
Pour effectuer une séparation des protéines
selon leur poids moléculaire, il est important que vos
protéines soient dénaturées (effet
chaotropique) et portent une charge. En effet, après
la séparation selon leur pI, vos protéines sont
dépourvues de charge nette, il est donc nécessaire
de leur en redonner une afin de pouvoir effectuer une nouvelle
électrophorèse. Le SDS porte une charge négative
et une queue hydrophobe. Environ deux molécules de SDS se
lient pour chaque acide aminé de la protéine. Par
conséquent, les protéines acquièrent une
charge nette négative fournie par les molécules de
SDS (attention il y a des exceptions, certaines protéines
ne fixent pas du tout le SDS).
|
|
|
|
|
|
|
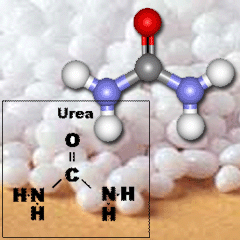
Mauvaise réponse. L’Urée.
Pas génial. L’urée est certes utilisée
pour dénaturer les protéines ou les solubiliser (en
détruisant les liaisons électrostatiques qui leur
donnent leur conformation), mais c’est un agent
chaotropique non-ionique. Par conséquent, et
contrairement au SDS (qui est un composé ionique), elle
n’ajoute pas de charge aux protéines (qui resteront
neutres et ne migreront pas). Vers bonne réponse.
A titre d’information, l’Urée est également
utilisée afin de dénaturer les acides nucléiques
(pour analyser le produit d’une réaction de
séquençage p.ex), contrairement au SDS.
|
Question : pourquoi n’avoir
pas rajouté le SDS avant l’isoélectrofocalisation
?
Votre gel polyacrylamide-SDS est donc prêt. Vous le collez
contre votre premier gel, perpendiculairement, afin de réaliser
la seconde électrophorèse (pour une illustration,
retournez au fichier de synthèse).
Vous branchez le courant et faites cette fois bien attention à
contrôler la durée de votre migration.
Questions : Au fait, vers quel pôle
migrent les protéines liées au SDS ? Et
durant l’isoélectrofocalisation, de quel côté
du gel avez-vous branché le pôle positif ?
Après 45 minutes, votre migration s’est bien passée
et maintenant, il vous faut visualiser vos protéines.
Etape 4 : Autoradiographie.
Dans cette étape vous allez enfin découvrir le
résultat de votre expérience.
Votre séparation des protéines par électrophorèse
en deux dimensions ayant été correctement réalisée,
vous devez maintenant détecter les protéines afin de
pouvoir comparer les protéomes de S. mutans selon les
différentes conditions testées (pH acide vs pH neutre
et culture planctonique versus biofilm).
Pour cela, devez-vous effectuer une coloration du gel avec du bleu
de coomassie (un colorant qui colore toutes les protéines,
tout comme le bromure d'éthydium, utilisé durant
l'expérience précédente, colorait tous les
acides nucléiques) ?
Pour vous aider, souvenez-vous que vous avez effectué un
marquage radioactif de vos protéines, mais que vous avez un
mélange de protéines marquées et non-marquées.
B. Emery. 21.11.05